Plasmid confirmation from colonies without overnight minipreps
This protocol describes a plate-parallel workflow for confirming plasmid constructs directly from bacterial colonies using micro-alkaline lysis, a two-stage SPRI cleanup, and Oxford Nanopore rapid barcoding. The front end is deliberately low-cost and enzyme-light: one colony is picked into one well, lysed with NaOH/SDS, neutralized with potassium acetate, clarified, depleted of high-molecular weight genomic DNA by a low-ratio bead step, and then captured at a higher bead ratio to enrich plasmid DNA. Each well is assigned a unique ONT rapid barcode during library prep, pooled, sequenced in real time, and analyzed by barcode-specific clone validation to call pass/fail, insert orientation, junction correctness, and structural discrepancies. Exact low-ratio and high-ratio bead settings are presented here as pilot starting points rather than final universal values.
Scope and expected performance
The purpose of Colony-to-Sequence is to compress the standard cloning verification loop from days to hours. Instead of growing overnight cultures and preparing minipreps before Sanger sequencing (or alternatively sending out for external ONT sequencing elsewhere), each colony is treated as a direct input. One colony is placed in one well, processed independently through lysis and cleanup, assigned one rapid barcode during library preparation, then pooled for one sequencing run. ONT’s plasmid sequencing protocol for SQK-RBK114.24/.96 explicitly supports multiplexing up to 96 plasmid samples, with
downstream analysis via wf-clone-validation to produce consensus sequences and clone-validation outputs.
For an initial pilot, a 24-plex workflow is suggested to be completed in roughly 2–3 hours from colony picking to analysis-ready FASTQ/BAM, with sequencing stopped once each barcode reaches a predefined coverage target rather than after a fixed time. A full 96-plex format is the scale target (if needed), but not the day-one guarantee. The critical front-end variables to be optimized are colony biomass plasmid DNA yield, minimizing genomic DNA shearing, and DoE-based tuning of the first and second bead ratios.
Materials and equipment
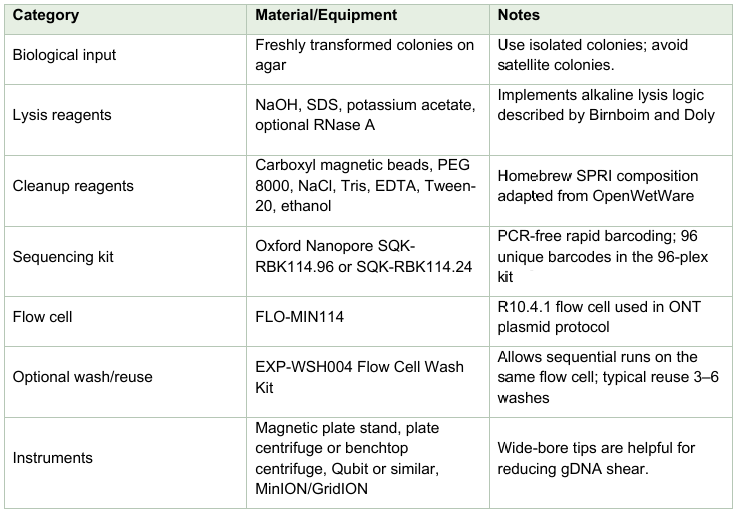
Homebrew DNA-binding SPRI bead mix
Adapted from Philippe Jolivet and Joe Foley (via OpenWetWare) who created a low-cost DNA-binding bead formulation containing 20% PEG 8000, 2.5 M NaCl, 10 mM Tris, 1 mM EDTA, 0.05% Tween-20, plus a washed carboxyl magnetic bead suspension.
The 50 mL formulation is:
- 3.582 mL nuclease-free water
- 25.00 mL of 5 M NaCl
- 0.50 mL of 1 M Tris
- 0.50 mL of 0.1 M EDTA
- 0.168 mL of 1 N HCl
- 20.00 mL of 50% PEG 8000
- 0.250 mL of 10% Tween-20
- 1.00 mL washed Sera-Mag-type bead suspension
Store the finished bead mix at 4 °C and mix thoroughly before use.
Prototype micro-alkaline lysis solutions
Use a freshly prepared NaOH/SDS lysis solution and a potassium acetate neutralization solution. The underlying principle is selective alkaline denaturation of high-molecular-weight chromosomal DNA while covalently closed circular plasmid DNA can be recovered after neutralization. Exact micro-scale volumes below are prototype starting conditions for pilot optimization.
protocol
Create a fixed plate map before picking colonies. Add 15 µL nuclease-free water or TE to each target well of a PCR plate or 96-well plate. Pick one colony into each well. Include at minimum one blank well, one known-correct positive-control colony, and one deliberate mixed-colony stress-control well. This plate map
will later be matched one-to-one to the ONT barcode assigned counterparts.
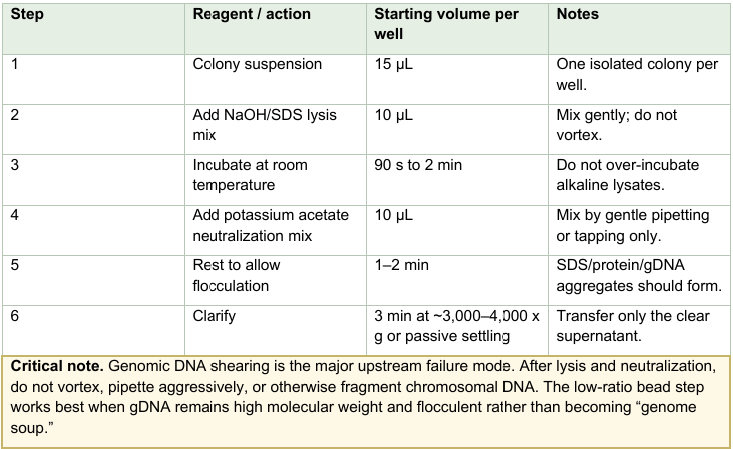
Micro-alkaline lysis
Dual-SPRI plasmid enrichment
Transfer 20 µL of clarified supernatant to a fresh plate. The first bead step is a depletion step for very large DNA and debris; the second bead step is a capture step for plasmid DNA. Because this selectivity depends on the local bead chemistry, these values should be piloted as a small matrix rather than treated
as final constants.
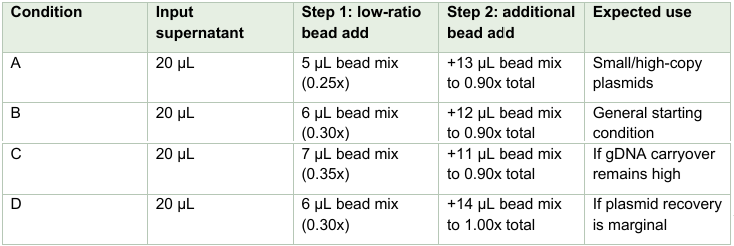
For the low-ratio step, add the bead mix, pipette gently to mix, incubate 2–3 minutes, place on a magnet until clear, and retain the supernatant. For the capture step, add the second bead volume directly to the retained supernatant, mix gently, incubate 2–5 minutes, magnetize, discard the supernatant, wash the beads twice with 100 µL of 70% ethanol, briefly air-dry, and elute plasmid DNA in 10–20 µL water or low salt buffer.
Minimum yield checkpoint
Measure DNA with Qubit or an equivalent fluorometric assay if possible. ONT’s plasmid sequencing protocol specifies 50 ng high-molecular-weight plasmid DNA per sample for SQK-RBK114 plasmid sequencing. As a practical starting rule, wells yielding at least 50 ng total eluate can move directly into rapid barcoding. Wells below this threshold can either be excluded from the run or routed to the optional rolling-circle amplification branch.
ONT rapid barcoding library preparation
Follow the current ONT plasmid sequencing protocol for SQK-RBK114.24 or SQK-RBK114.96. The essential layout is: assign one unique rapid barcode to each colony-derived sample, tagment the plasmid DNA with that barcode, pool the barcoded wells, perform the kit cleanup, attach rapid sequencing adapters, then load on FLO-MIN114. ONT states that the 96-plex kit contains 96 unique barcodes, supports up to 96 libraries, and uses a PCR-free transposase workflow in which barcoded samples are SPRI cleaned and pooled before adapter addition.
Optional rescue protocol: rolling-circle amplification
If certain wells repeatedly fail the 50-ng input checkpoint or produce poor barcode balance, add a rescue set (branch) of samples to be amplified based on phi29 DNA polymerase rolling-circle amplification. we have showed that primed RCA can amplify plasmid DNA from single colonies or plaques by roughly
10,000-fold in a few hours, providing direct sequencing templates without lengthy growth. In this protocol, RCA is best treated as a conditional rescue step after the low-ratio/high-ratio cleanup rather than as the default path for every colony. Any commercial or in-house phi29 RCA implementation can be used according to its current instructions. I suggest New England Biolabs phi29-XT RCA Kit (E1603S).
Sequencing run design and stopping criteria
Start the sequencing run in MinKNOW with live basecalling and barcode demultiplexing enabled. ONT documents say that MinKNOW currently uses Dorado for both basecalling and barcode demultiplexing and can split reads into barcode-specific folders in real time as the run progresses. ONT’s plasmid protocol
recommends 12 hours for new users, although it explicitly notes that shorter run times may be sufficient. For this workflow, the defensible strategy is not to promise a fixed 5–15 minute run, but to stop the run automatically once every barcode reaches a predefined clone-validation coverage target.
The wf-clone-validation method sets the default assembly coverage to 60x and states that, because the workflow performs three assemblies, it targets approximately 3x the required coverage during subsampling, A practical heuristic is therefore to target about 180x total mapped bases per barcode. For a 7 kb
plasmid, that corresponds to approximately 1.26 Mb of mapped plasmid sequence for each well. In a tuned 24-plex run this may be reached in tens of minutes; in a full 96-plex format, longer run times should be expected.
Data analysis and pass/fail logic
Use MinKNOW live output or post-run demultiplexing to create one barcode-specific read set per colony, Run each barcode set through EPI2ME wf-clone-validation, optionally providing: (i) an approximate plasmid size, (ii) a full plasmid reference FASTA, (iii) an insert reference FASTA, (iv) primer sequences for
locating inserts, and (v) a host reference if host-read filtering is desired. The workflow performs de novo plasmid reconstruction, full-assembly comparison, per-base quality scoring, insert location by primers, and annotation.
Define the pilot pass/fail logic as follows: PASS if the final assembly length is within an acceptable band of the expected plasmid size, the expected insert is present, junctions are correct, orientation matches the design, and no large deletion, duplication, or rearrangement is detected relative to the expected construct.
FAIL if assembly size is strongly discordant, insert or junctions are incorrect, or the per-sample data are insufficient for a confident call. A mixed-colony or cross-contamination warning can be implemented as a custom QC overlay by inspecting the proportion of reads that disagree with the dominant assembly or by reviewing variant fractions against the expected reference.
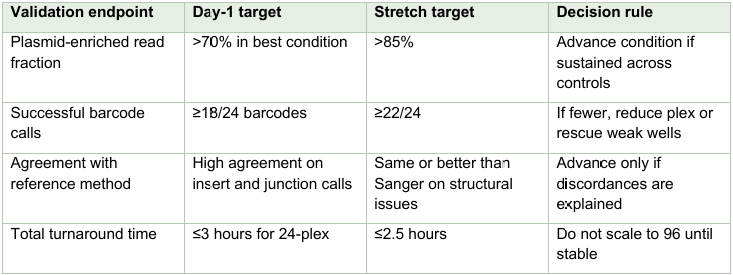
Pilot validation criteria
Begin with a 24-colony pilot rather than 96 colonies. Include at least three plasmid classes: a 3–5 kb high copy plasmid, a 7–10 kb moderate plasmid, and one larger or lower-yield construct that represents a stressful but realistic use case. Split colonies across the bead-ratio conditions in Dual-SPRI plasmid enrichment (see above). Primary readouts should be total DNA yield, fraction of reads mapping to plasmid versus host, percentage of barcodes producing a complete clone-validation report, and benchmarking against conventional miniprep plus Sanger or miniprep plus orthogonal plasmid sequencing.
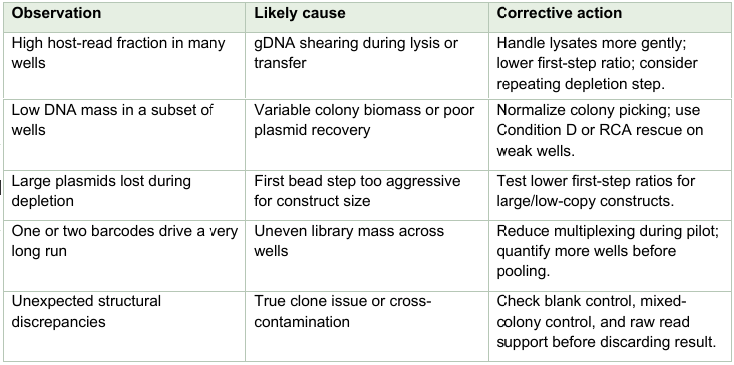
Troubleshooting and failure modes